Hello,
I want to make HPLC of Ru dye chelats (RuL2(NCS)2, L=2,2'Bipyridyl-4,4'-Dicaroxylat). I use reversed phase column RP8. My mobile phase is Acetonitrile and H2SO4 (0,0001mol). I have tried to solve the dye in Ethanol, Methanol and NaOH. I use Dioden Array Detector. My problem is, that I get two peaks at different times, the first at 0.7 min, the second at 4min. Both peaks posses the same, or almost the same absorbtion spektrum with three maxima at 300, 340 and 500nm. I have already tried to change the column, but it doesn't help. Can you help me?
![]()
![]()
![]()
![]()
![]() By Chris Pohl on Monday, February 3, 2003 - 01:30 pm:
By Chris Pohl on Monday, February 3, 2003 - 01:30 pm:
Two peaks man,
Are the two peaks bridged (i.e. does it completely return to baseline in between the two peaks)? If so, there's a good chance your complex is falling apart. I'm not familiar with the spectral properties of the ligand, so I can't guess as to which peak is which. Does 2,2'bipyridyl-4,4'-dicarboxylate absorb at the specified wavelengths?
Why are you working at low pH? It would be safer to utilize higher pH in order to prevent dissociation of the complex on column.
![]()
![]()
![]()
![]()
![]() By two peaks man on Monday, February 3, 2003 - 04:03 pm:
By two peaks man on Monday, February 3, 2003 - 04:03 pm:
I have already tried a mobile phase: acetonitrile with destilled water, so the pH was 7, but it didn't help. my chromatogram looks like this: 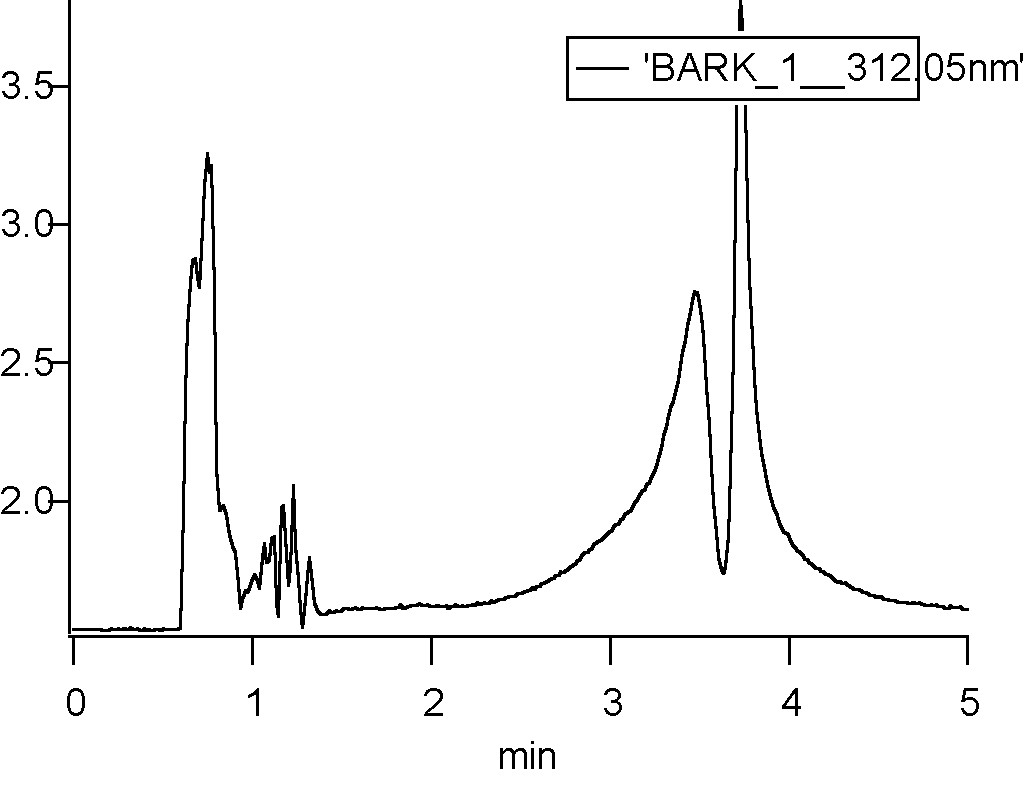
![]()
![]()
![]()
![]()
![]() By two peaks man on Monday, February 3, 2003 - 04:06 pm:
By two peaks man on Monday, February 3, 2003 - 04:06 pm:
I forget to mention that the peaks with same spektrum are the three highest peaks in my chromatogram
![]()
![]()
![]()
![]()
![]() By Anonymous on Tuesday, February 4, 2003 - 11:17 am:
By Anonymous on Tuesday, February 4, 2003 - 11:17 am:
It looks like your peak is splitted by quite symmetrical negative peak.
![]()
![]()
![]()
![]()
![]() By Chris Pohl on Tuesday, February 4, 2003 - 12:56 pm:
By Chris Pohl on Tuesday, February 4, 2003 - 12:56 pm:
Two peaks man,
From the look of it, it appears that your complexes falling apart. I would suggest that you work with a buffered eluent (using an acetate buffer) at around pH 4-5. If this isn't enough to stabilize things, you may need to add excess ligand to your sample. When in doubt, it's always beneficial to that excess ligand to your eluent but considering your ligand will absorb strongly in the ultraviolet, this option is probably not available to you.
![]()
![]()
![]()
![]()
![]() By HW Mueller on Wednesday, February 5, 2003 - 12:45 am:
By HW Mueller on Wednesday, February 5, 2003 - 12:45 am:
My reply from yesterday disappeared, so here another trial:
To me it also looks like a negative peak (~3.7min) superimposed on a very badly chromatographing peak (wrong conditions or reaction as Chris proposed).
What is your dead time, your flow, and column dimensions?
![]()
![]()
![]()
![]()
![]() By two peaks man on Wednesday, February 5, 2003 - 03:13 am:
By two peaks man on Wednesday, February 5, 2003 - 03:13 am:
My flow time is 1.5 ml/min, my columns size is 250*4 mm, RP8 column Eurospher 100.
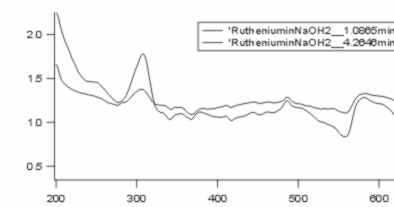
I've just made another measure, now it looks a little bit different, I show you my chromatogram and the spektrums of the two peaks. This time I solved my complex in NaOH. I've again two peaks.
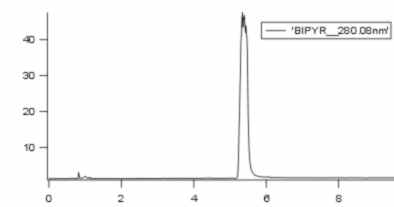
I've also tried to make a HPLC with the Ligand (Bipyridil). Bipyridil comes at 5min. So if my compex is falling apart, the first peak at 1min should be the complex, and the second part at 4min should be the ligand(bipyridil). Yes Chris, the Ligand absorbs in UV.
And here are the chromatograms in this order 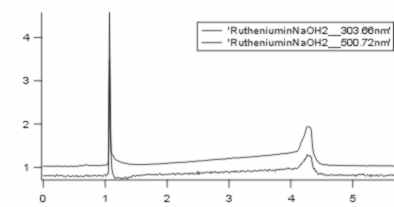
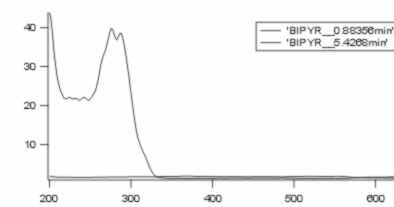
![]()
![]()
![]()
![]()
![]() By two peaks man on Wednesday, February 5, 2003 - 03:27 am:
By two peaks man on Wednesday, February 5, 2003 - 03:27 am:
The order of the pictures is :
1. chromatogram of Ru dye in NaOH
2. spektrum of the two peaks of the previous chromatgram
3. chromatogram of Bipyridyl
4. spektrum of Bipyridyl
![]()
![]()
![]()
![]()
![]() By HW Mueller on Thursday, February 6, 2003 - 12:27 am:
By HW Mueller on Thursday, February 6, 2003 - 12:27 am:
The dead time (tm or to) of your column should be around 1min under the info you gave(if the assumptions for calc of tm applies here). Your first peak in the first chromatogram is then probably not retained, ie, not chromatographed. The second peak is still very badly chromatographed, though the neg "peak" is gone. You should always take a spectrum of the baseline, preferably at several points, otherwise you cannot assign a spec to peaks. Your bipyridyl peak is badly split, too much for the detector? inhomogeneous??, nervous detector?
Looks like you have to play around quite a bit with your chromatography. No refs?
![]()
![]()
![]()
![]()
![]() By two peaks man on Thursday, February 6, 2003 - 03:20 am:
By two peaks man on Thursday, February 6, 2003 - 03:20 am:
Dear HW Mueller,
unfortunatly I've got no refs. I think that the first peak is not retained, too. But what could be the reason for it ? Why is a part of substance retained and another part not?
![]()
![]()
![]()
![]()
![]() By HW Mueller on Thursday, February 6, 2003 - 07:27 am:
By HW Mueller on Thursday, February 6, 2003 - 07:27 am:
Presently my guess is that your spectra are due to a dirt continuum (your baseline) and that you do not know where your complex is. Do you have a independent spectrum of the Ru-complex? It may also be possible that your complex, or degradation products thereof, smear out over the entire column.
If the two peaks are really the same substance it could mean that you have a channel through the column. What happens if you collect the peaks and reinject them?
![]()
![]()
![]()
![]()
![]() By two peaks man on Thursday, February 6, 2003 - 12:43 pm:
By two peaks man on Thursday, February 6, 2003 - 12:43 pm:
I have tried to change the column, but the result was the same. I have an independent spektrum of the substance, and its almost the same than the spektrum of my peaks. The spectrum of the baseline is rather flat, and has not the three maxima like my dye.
I've never tried to colect the peaks and reinject my substance again, that is a good idea, I am going to try it.
Thank you
Markus
![]()
![]()
![]()
![]()
![]() By HW Mueller on Thursday, February 6, 2003 - 11:28 pm:
By HW Mueller on Thursday, February 6, 2003 - 11:28 pm:
Of course, an obvious possibility is also that you have two isomers or just two complexes. The reinjection should be done at different times (there may be an interchange) and/or workup conditions.
![]()
![]()
![]()
![]()
![]() By Andreas Neumaier on Friday, February 7, 2003 - 01:21 am:
By Andreas Neumaier on Friday, February 7, 2003 - 01:21 am:
Dear two Peaks man,
Once I had similar problems with a substance that tends to build complexes und every time I used H2SO4 as acid in the mobile phase the little racker shots right through any column (even Perfluorphenyl). When I changed to acetic acid or TFA I got normal retention (for Perfluorphenyl retention was about 3x higher then compared to any other column).
As far as I could explane the mechanism to me, the big H2SO4-molecul enveloped the substance and made it real polar on the outside of the complex, so it wasn't retained at all on any column I used. Think that's what happened to you too.
Did you tried to change your acid in the mobile phase from H2SO4 to TFA or acetic acid? (See also comment from Chris Pohl, 04.02.03)
Good luck, Andreas