I report related impurities by % Area. My problem is by changing the detector % impurities value get changed for example if total impurities reported 2.35% same sample analysed with other detector it shows total impurities 1.98%.
Can detector make so much different?
How to find out difference between two detector which effecting the results?
![]()
![]()
![]()
![]()
![]() By Russ on Monday, December 1, 2003 - 05:55 am:
By Russ on Monday, December 1, 2003 - 05:55 am:
What detector are you using, UV, RI, MS?
![]()
![]()
![]()
![]()
![]() By Anonymous on Tuesday, December 2, 2003 - 05:16 am:
By Anonymous on Tuesday, December 2, 2003 - 05:16 am:
Hi Russ
I am using Variable wavelegth Detector (UV-Visible Detector)
![]()
![]()
![]()
![]()
![]() By readski on Tuesday, December 2, 2003 - 05:17 am:
By readski on Tuesday, December 2, 2003 - 05:17 am:
Are you running these comparisons on the same column and pump and just switching detectors?
![]()
![]()
![]()
![]()
![]() By Anonymous on Tuesday, December 2, 2003 - 05:39 am:
By Anonymous on Tuesday, December 2, 2003 - 05:39 am:
I've seen differences between a photo diode array (PDA)and a single/multi wavelength detector. The single wavelength detector will give you cleaner chromatography at the noise level than the PDA. The PDA will scan a range of wavelengths, say from 210 to 400 nm, then the user will extract the desired wavelength from the scan so between those two types of UV detectors, the single/multi wavelength detector is the more accurate at the impurity level. However, if you're comparing two of the same detectors, lamp hours/age can produce different results. Look at the when the detectors were last PM'd. Hope this helps.
![]()
![]()
![]()
![]()
![]() By Andreas Neumaier on Tuesday, December 2, 2003 - 08:59 am:
By Andreas Neumaier on Tuesday, December 2, 2003 - 08:59 am:
First:
Chromatograms aren't meassured at the exact wavelenght which is stated from the software.
They are meassured in a range from wavelength minus bandwith to wavelength plus bandwith (when bandwith is reported as +/- x nm). For VWD the bandwith is fixed (read the manuals for exact data), for DAD there are normally fixed bandwith steps, which influence the interpolated wavelength the software is showing you.
=> So different detectors must give you different absorbance at the "same" wavelength.
Second:
Wavelength accuracy has more or less influence on the meassured wavelength. Most detectors can meassure the wavelength accuracy (normally at 486.0 and 656.1 nm).
The quality of the lamp has influence on the wavelength accuracy.
=> Even with two identical detectors you can have different absorbance at the "same" wavelength.
You can try to reduce the influence fron this two points by using a wavelength where all spectras of your compounds has over some nm no raise or fall of absorbance (means the spectra of every peak is meassured at a plateau).
I try to add two pictures of the same spectra as an example.
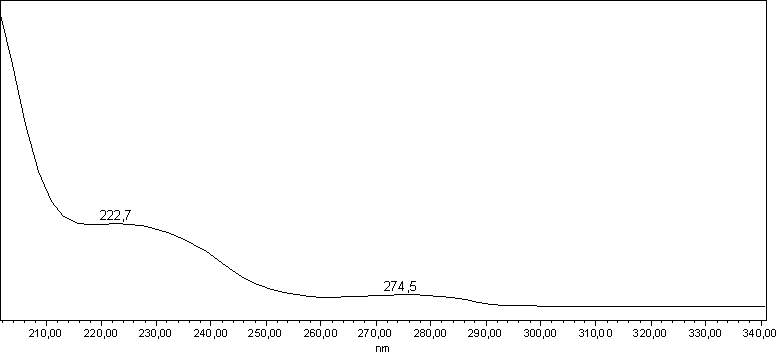
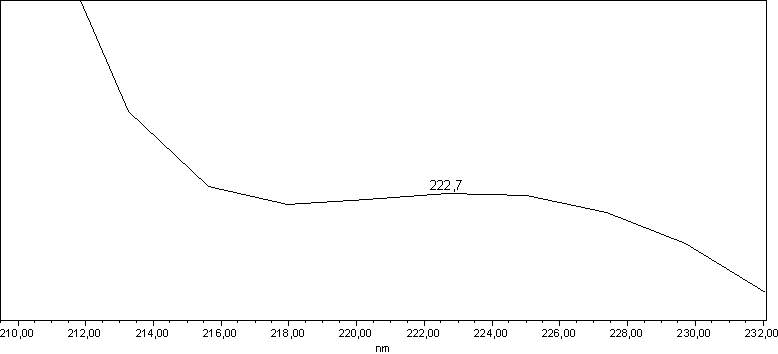
![]()
![]()
![]()
![]()
![]() By Andreas Neumaier on Tuesday, December 2, 2003 - 09:03 am:
By Andreas Neumaier on Tuesday, December 2, 2003 - 09:03 am:
The wavelength of 222 nm should result in minimal differences for area% with different detectors.
At 240 nm different detectors will most likely report different area%
![]()
![]()
![]()
![]()
![]() By HW Mueller on Tuesday, December 2, 2003 - 11:40 pm:
By HW Mueller on Tuesday, December 2, 2003 - 11:40 pm:
All UV spectrometers, detectors.. have to be calibrated and checked with standards. If you then stay within the linear range, why should there be a difference in sample measurement?
If there still were diffs one could forget about quantitation with UV.
Also, I must be living in a different world. A 0.37% diff. at 2%? Measurement to a 1/100%? Displacement of a peak marker by a millimeter can cause a few % area change, easily.
![]()
![]()
![]()
![]()
![]() By readski on Wednesday, December 3, 2003 - 05:20 am:
By readski on Wednesday, December 3, 2003 - 05:20 am:
HW- Great observation regarding % difference
But we may have overlooked the following...
What if the detectors are set to different sensitivities? Wouldn't this be a possible reason for a change (even though it is only 0.37%) in total % impurities?
![]()
![]()
![]()
![]()
![]() By HW Mueller on Wednesday, December 3, 2003 - 06:37 am:
By HW Mueller on Wednesday, December 3, 2003 - 06:37 am:
readski, via the integration setting, maybe, if you have a sensitivity setting at the integrator output?
![]()
![]()
![]()
![]()
![]() By Anonymous on Wednesday, December 3, 2003 - 07:44 am:
By Anonymous on Wednesday, December 3, 2003 - 07:44 am:
Hi Readski
Yes I am running on the same column and same mobile phase (For your information Chromatogram pattren is same, I mean number of peak and separation is same) but pump and detector is different.
Hi HW Mueller
Yes I agree with you but applying all integration parameters, above said difference is still there.
I calibrated both detector before analysis and it was found satisfactory.
Hi Readski
Detector are set to same sentivities.
Thanks to you all for your information.
I forget to tell you one things in my previous question that chromatogram integration was done with different sofware one with Winchrom software which reports 2.35% total impurities and Chromeleon software which reports 1.98% total impurities. I quantify all the peak applying best combination of integration parameters? Even software integration can make this difference?
Your suggestion will be highly appreciated.
![]()
![]()
![]()
![]()
![]() By HW Mueller on Wednesday, December 3, 2003 - 11:51 pm:
By HW Mueller on Wednesday, December 3, 2003 - 11:51 pm:
Why don�t you play a bit with your integration parameters (for instance, changing slope or euivalent), you will see how this effects results.
![]()
![]()
![]()
![]()
![]() By Beppe on Thursday, December 4, 2003 - 12:12 am:
By Beppe on Thursday, December 4, 2003 - 12:12 am:
From my experience, the factors that can lead to differences in area % are :
1) bandwith effect (as explained by Andreas)
2) effects on the main peak : does it saturate one detector ad not the other ? is the main peak shape identical (tailing, broadness ... differences may also come from pump, various dead volumes)
3) effect of some "hidden" parameters of the couple detector/signal numerization circuits like sampling rate, noise filtering/time constant, high level signal cut (on the main peak)
4) integretion parameters : do not only look at their influence on impurity peaks area; they may have a larger impact on how the main peak area is computed.
![]()
![]()
![]()
![]()
![]() By HW Mueller on Thursday, December 4, 2003 - 11:31 pm:
By HW Mueller on Thursday, December 4, 2003 - 11:31 pm:
Beppe,
you mean wavelegth bandwidth in your 1)? The Beer-Lambert Law is independent of bandwidth, so relative values like the mentioned % should be independant of bandwidth. If your mashines are calibrated even "absolute" values (concentrations) will not depend on bandwidth.
Now on 3): One almost has to try to misadjust the apparatus to get an effect here.
![]()
![]()
![]()
![]()
![]() By Russ on Friday, December 5, 2003 - 05:44 am:
By Russ on Friday, December 5, 2003 - 05:44 am:
HW, maybe I am not understanding something but if you are reporting results as area %, and if the spectra of the compounds making up the total area are not the same, couldn't instrument differences and bandwidth have an effect? For example, consider two compounds A and B. Compound A has a spectrum that is "flat" at the wavelength monitored. The spectrum for B has a sharp slope at the monitored wavelength. Obviously if the instruments set the wavelengths within very narrow ranges, this difference would be very small. But within the error range of most instruments, it would seem that, under these conditions, you could get different results from different instruments, or even from the same instrument depending on the instruments internal "calibration" (that is if a "true" 254 nm is called 252 on one day and 256 on the next). Like I said, I may be overlooking something.
![]()
![]()
![]()
![]()
![]() By Anonymous on Friday, December 5, 2003 - 02:44 pm:
By Anonymous on Friday, December 5, 2003 - 02:44 pm:
Hans,
I thought Beers law was only true for a single wavelength. I am pretty sure different bandwidth settings can affect linearity and quantification.
![]()
![]()
![]()
![]()
![]() By HW Mueller on Monday, December 8, 2003 - 12:06 am:
By HW Mueller on Monday, December 8, 2003 - 12:06 am:
Beer�s... law is independent of wavelength. The only problem you can encounter regarding bandwidth is if the B-L law does not apply (nonlinearity). If you increase the wavelength bandwidth you get more light through, both with and without sample in the path, the ratio (Io/I) stays the same.
![]()
![]()
![]()
![]()
![]() By Anonymous on Friday, December 12, 2003 - 02:02 pm:
By Anonymous on Friday, December 12, 2003 - 02:02 pm:
Beer's law is independent of wavelength but its derivation is based on monchromatic incident light, which is never truly achieved.
On detectors like the Agilent 1100 DAD where you can change the slit width and bandwidth, this can affect linearity.
http://www.biochem.purdue.edu/~courses/undrgrad/221/wwwboard/handouts/overheads/lect_15/03lecture15oh.pdf?cfC0A89682=VEhFUkFWQU5DRVxUTWl6dWthbWk6dGhlcmF2YW5jZTobahSrD69cDL7eYCQAR44C
![]()
![]()
![]()
![]()
![]() By HW Mueller on Monday, December 15, 2003 - 04:22 am:
By HW Mueller on Monday, December 15, 2003 - 04:22 am:
Last anon: Your ref. talks about absolute concentrations (obviously if more light goes through due to enlarging the slit your concentration seems to go down). That�s comparing apples to elephants (not pears). It was mentioned above that you have to calibrate or adjust your apparatus and then stick with that if you want to compare two or more values. Or in HPLC with internal standard you do this practically automatically. Again, you can not change the parameters after you tought your mashine how much light passage is associated with a given concentration. If you do change parameters you have to readjust or recalibrate. Also again, if it were not so UV mashines would be useless.
![]()
![]()
![]()
![]()
![]() By HW Mueller on Tuesday, December 16, 2003 - 05:50 am:
By HW Mueller on Tuesday, December 16, 2003 - 05:50 am:
OK, reviewing this once more it seems that I garbled some of my explanations. The absorption coefficient has to be, of course, used only for wavelengths with the same absorption. So to get at the concentration (absolutely) of a substance, if a UV spectrometer is used, one usually sets it at the wavelength at which the absorption coefficient (a) was used (if not you also have to calibrate here). Now in HPLC we make relative measurements (unless you want to predict area, as discussed in an old chain) so that even if one "catches" wavelength with different "a" (wide slit. sharp spectrum), etc., the effect is cancelled.